One of the issues I discussed at our SBM workshop was something I’ve written before, namely the “methodolatry” that sometimes infests evidence-based medicine (EBM), “Methodolatry” has been defined as the profane worship of the
randomized clinical trial (RCT) as the only valid method of clinical investigation, and it’s a symptom of the way that EBM relegates basic science knowledge, even well-established principles of science that show that something like, say, homeopathy or reiki is impossible under the current understanding of physics, chemistry and biology. However, never let it be said that RCTs aren’t actually important in SBM. Our problem with how EBM worships them derives from how it even bothers to do trials in the first place of modalities that can best be described by Harriet Hall’s brilliant appellation,
Tooth Fairy Science. However, these days RCTs are widely perceived to have a serious problem.
They have become so expensive to do and there have been so many failures of drugs that looked promising to show efficacy in clinical trials that some have even questioned whether there is something fundamentally wrong with how we do clinical trials now. Some even ask, as the title of an article by Clifton Leaf that appeared in the New York Times over the weekend,
Do Clinical Trials Work?
It begins with the story of Avastin in brain tumors. I’m sure that Eric Merola will likely jump all over this, given how he tried to use the example of Avastin being approved for glioma on fast track approval that used phase II trials as the basis for doing so as an argument for why antineoplastons should be approved by the FDA. Or maybe he won’t. Here’s why.
The story explains that there were two single-arm trials of adding Avastin to glioma therapy in which the tumors “shrank and the disease seemed to stall for several months when patients were given the drug.” Then Clifton points out the results of the randomized clinical trial presented at the American Society of Clinical Oncology (ASCO) meeting a month and a half ago:
But to the surprise of many, Dr. Gilbert’s study found no difference in survival between those who were given Avastin and those who were given a placebo.
Disappointing though its outcome was, the study represented a victory for science over guesswork, of hard data over hunches. As far as clinical trials went, Dr. Gilbert’s study was the gold standard. The earlier studies had each been “single-arm,” in the lingo of clinical trials, meaning there had been no comparison group. In Dr. Gilbert’s study, more than 600 brain cancer patients were randomly assigned to two evenly balanced groups: an intervention arm (those who got Avastin along with a standard treatment) and a control arm (those who got the latter and a placebo). What’s more, the study was “double-blind” — neither the patients nor the doctors knew who was in which group until after the results had been assessed.
The centerpiece of the country’s drug-testing system — the randomized, controlled trial — had worked.
This study could certainly be taken as evidence supporting a position that we shouldn’t approve drugs based on single-arm phase II clinical trials, even under fast track. It is indeed a very good example of how promising phase II clinical trial results are not always validated when the bigger and more rigorous phase III RCTs are performed. In one way, it is a good thing. Negative results, be they experimental or clinical trial, are just as important in science as positive results, if not more so. In another way, however, it’s a bad thing because, as the NYT article points out,
“doctors had no more clarity after the trial about how to treat brain cancer patients than they had before.” A seemingly promising addition to the armamentarium against a deadly cancer that has too few effective treatments was shown not to work in an RCT that was designed to be, more or less, definitive. However, the key thing to remember about such an RCT is that it is looking at populations of patients.
Overall, there was no difference in overall survival between the control and Avastin group, but that doesn’t necessarily mean that Avastin is useless against glioma.
Indeed, as someone who’s been studying angiogenesis and how to target it therapeutically in cancer since the heady days of the late 1990s, when findings by Judah Folkman and other pioneers in this field led to headlines in the lay press like “The Cure for Cancer” and it really did look as though the discovery that inhibiting angiogenesis produced dramatic results and outright cures in preclinical rodent models of cancer. Over the years, the study of angiogenesis has been gradually de-emphasized in my research, correlating inversely with the rise of other interests, but I do have a small project in targeting tumor-induced angiogenesis still ongoing and hope to publish on it before the end of the year. In any case, reality shut down those heady days, as it became clear that Avastin and other antiangiogenics were not as nontoxic in humans as they were in mice, nor were they nearly as effective. Still, it is clear that Avastin has contributed to significant increases in median survival in a number of tumor types, such as colorectal cancer. However, overall it’s hard not to conclude that antiangiogenic therapy has been, by and large, a disappointment, if only because the hype and hope were so sky-high 15 years ago. Rare indeed would have been the treatment that could have lived up to such expectations when tested in RCTs.
One thing that has been apparent for quite some time is that there appears to be a subset of patients who have remarkable responses to Avastin. Many oncologists get this feeling anecdotally, even if they don’t have evidence, and evidence has popped up in clinical trials. Assuming this is true, while it might not now make sense to treat all or most glioma patients with Avastin, it might very well make sense to treat that subset who have such dramatic responses if we could identify them beforehand. There’s the rub, though. We can’t, and Leaf points this out:
Some patients did do better on the drug, and indeed, doctors and patients insist that some who take Avastin significantly beat the average. But the trial was unable to discover these “responders” along the way, much less examine what might have accounted for the difference. (Dr. Gilbert is working to figure that out now.)
Indeed, even after some 400 completed clinical trials in various cancers, it’s not clear why Avastin works (or doesn’t work) in any single patient. “Despite looking at hundreds of potential predictive biomarkers, we do not currently have a way to predict who is most likely to respond to Avastin and who is not,” says a spokesperson for Genentech, a division of the Swiss pharmaceutical giant Roche, which makes the drug.
That we could be this uncertain about any medicine with $6 billion in annual global sales — and after 16 years of human trials involving tens of thousands of patients — is remarkable in itself. And yet this is the norm, not the exception. We are just as confused about a host of other long-tested therapies: neuroprotective drugs for stroke, erythropoiesis-stimulating agents for anemia, the antiviral drug Tamiflu — and, as recent headlines have shown, rosiglitazone (Avandia) for diabetes, a controversy that has now embroiled a related class of molecules. Which brings us to perhaps a more fundamental question, one that few people really want to ask: do clinical trials even work? Or are the diseases of individuals so particular that testing experimental medicines in broad groups is doomed to create more frustration than knowledge?
While it’s an excellent point that we don’t have predictive biomarkers (say, something in the blood we could measure) that tell us which patients are most likely to respond to Avastin (or most other drugs), Leaf seems to be indulging in a false dichotomy. Just because we don’t have predictive biomarkers for various drugs does not imply that clinical trials don’t work. Very clearly, they do. The problem is that they have limitations, and one of those limitations is that, without predictive biomarkers, we have no choice but to test the drug in a controlled population and see if there is a difference between control and the treated population that can be observed on a population level. The smaller the difference, the harder it is to detect and the more patients are needed to detect it. That’s why we need and want predictive biomarkers in the first place.
Worse, even the biomarkers we have are nowhere near 100% predictive. Let’s take a look at the prototypical targeted therapy, arguably the oldest targeted drug of all, Tamoxifen, which blocks estrogen activity. It is only used in tumors that make the estrogen receptor and are therefore presumed to be estrogen-responsive (i.e., estrogen stimulates them to grow). I remember a talk by the director of the Cancer Institute of New Jersey at the time I worked there, William Hait, who pointed out that Tamoxifen is effective in ER(+) cancers about 50% of the time. Around 70% of breast cancer is ER(+), and that means that if you treat all patients with breast cancer with Tamoxifen, you will see responses only 35% of the time, whereas if you treat only ER(+) cancers you will see responses 50% of the time. Another example is Herceptin, which targets amplified HER2 in breast cancer. Even though it is a targeted drug, it is effective against approximately 30% of HER2(+) cancers. Now, approximately 30% of breast cancers are HER2(+), which means that if you treat all comers with Herceptin, it will only be effective 0.3 x 0.3 = 0.09 (9%) of the time, but if you treat only HER2(+) cancers it should be effective 30% of the time. There are other examples he gave us. Taxol, for instance, is effective in 75% of breast cancer with p53 mutations. Since approximately 50% of breast cancers carry p53 mutations, if you treat all comers with Taxol you will get responses around 37.5% of the time, whereas if you treat only cancers with p53 mutations you should expect a 75% response rate. Of course, a 37.5% response rate is good enough that pretty much everyone with breast cancer who needs chemotherapy will get a Taxane, but you get the idea.
Now here’s where the devil is. These biomarkers that I’ve described are crude, and not even that predictive. But what, if anything, is better? That’s the problem, and that’s where most articles like this break down. They do an excellent job of identifying the problems with clinical trials, and there’s no doubt that Clifton Leaf does just that. None of these problems discussed in his NYT article are unfamiliar to most clinicians and clinical investigators, particularly in cancer. However, one notes that he has a book out entitled The Truth In Small Doses: Why We’re Losing the War on Cancer — and How to Win it. Personally, I hate that meme of “we’re losing the war on cancer,” because it’s not a war, and whether or not we’re “losing” depends on what your vision of “victory” is and how fast we can win the war. As I’ve pointed out many times, particularly around the 40th anniversary of Richard Nixon’s declaration of “war on cancer,” what do you expect in 40 years, given that the amount of resources we pour into this “war” are minuscule compared to what we spend on other things, such as—oh, you know—actual war? How much progress can we realistically expect in 40 years given that investment, the incredible complexity of cancer, and cancer’s ability to out-evolve almost anything we have as yet been able to throw at it. Clifton Leaf is a cancer survivor; so I can totally understand his frustration. However, that doesn’t stop his use of that tired old meme from irritating me. I’ll stop whining about that particular pet peeve of mine right now, but as everyone knows I do so love a good whine. Sorry.
My pet peeve aside, what can we do better? Most of us in oncology believe that
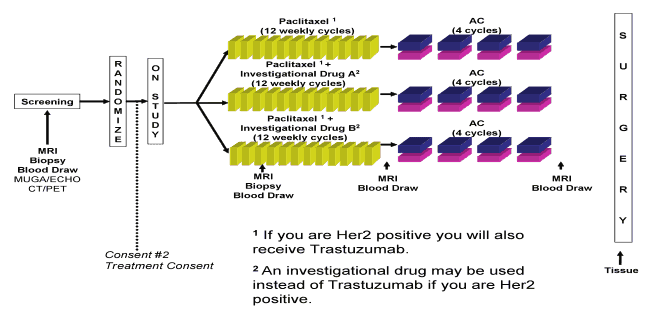
the answer will likely come down to personalized medicine based on the genomic profile of each cancer, but how to get from the enormous amount of data from genomic studies of various cancer to actual validated treatments is not at all clear at this stage (other knowing that Stanislaw Burzynski’s doing it wrong). Right now, personalized medicine has a lot of promise but has even more hype with little or nothing as yet in the way of concrete results that clearly benefit patients. Many have been the ideas to overcome these problems and validate genomic-based personalized medicine. Leaf actually mentions an interesting one: The I-SPY2 TRIAL (Investigation of Serial Studies to Predict Your Therapeutic Response with Imaging And moLecular Analysis 2). (Whew, what a name!) It’s a very interesting prototype of how clinical trials might be done in the future, and if it works I can see a lot more trials like this:
The I-SPY 2 TRIAL (Investigation of Serial Studies to Predict Your Therapeutic Response with Imaging And moLecular Analysis 2) is a clinical trial for women with newly diagnosed locally advanced breast cancer to test whether adding investigational drugs to standard chemotherapy is better than standard chemotherapy alone before having surgery. The treatment phase of this trial will be testing multiple investigational drugs that are thought to target the biology of each participant’s tumor. The trial will use the information from each participant who completes the study treatment to help decide treatment for future women who join the trial. This will help the study researchers learn more quickly which investigational drugs will be most beneficial for women with certain tumor characteristics. The I-SPY 2 TRIAL will test the idea of tailoring treatment by using molecular tests to help identify which patients should be treated with investigational drugs. Results of this trial may help make investigational drugs available to more women in the future.
The beauty of this trial is that it uses Bayesian analysis of responses to have the trial, in effect, evolve in response to what is found at earlier stages. My main quibble with the study is that it requires that all subjects undergo pretreatment breast MRI before surgery, which has a tendency to upstage women through the Will Rogers effect and thus result in more mastectomies. I understand that the trial investigators probably wanted advanced imaging to follow tumor response and that MRI can also show blood flow and therefore measure tumor angiogenesis, but I always worry when I see a design like this one, that it might promote unnecessary mastectomies. On the other hand, the inclusion criteria require a tumor that is 2.5 cm in diameter or greater so perhaps this will be less of a problem. That quibble aside, as Leaf describes, it is an intriguing design and it does evolve based on previous results:
In fact, a breast cancer trial called I-SPY 2, already under way, may be a good model to follow. The aim of the trial, sponsored by the Biomarkers Consortium, a partnership that includes the Foundation for the National Institutes of Health, the F.D.A., and others, is to figure out whether neoadjuvant therapy for breast cancer — administering drugs before a tumor is surgically removed — reduces recurrence of the disease, and if so, which drugs work best.
As with the Herceptin model, patients are being matched with experimental medicines that are designed to target a particular molecular subtype of breast cancer. But unlike in other trials, I-SPY 2 investigators, including Dr. Berry, are testing up to a dozen drugs from multiple companies, phasing out those that don’t appear to be working and subbing in others, without stopping the study.
Here’s the design (more details can be found
here and
here, and some of the investigational drugs tried can be found
here):
The difficult part of the study, of course, is designing the algorithms by which drugs are swapped out as they appear not to be working. If these decisions are made willy-nilly, then this trial would be no better than what Burzynski does (i.e., making simplistic guesses). However, there is a sophisticated analysis and algorithm by which treatment decisions are made. It does have to be remembered, though, that, although I-SPY2 does represent personalized medicine, it is not yet full genomic medicine. Most of the biomarker tests used are biomarkers that already exist, and the additional biomarkers measured will not affect patient treatment. This part of the trial is for discovery of biomarkers, not validation.
The bottom line
I’ll be watching the progress of I-SPY2 closely, because it’s a new kind of clinical trial. Whether it will succeed in improving the success of the followup clinical trials of agents identified through I-SPY remains to be seen, as it also remains to be seen whether it will speed up the pace of discovery. I’m probably less hopeful than Clifton Leaf, but that doesn’t mean I’m not hopeful.
So do clinical trials work? It depends on what you mean by “clinical trials” and “work.” I would argue that they do, in fact, still work in that they are still the best method we have to determine whether science-based therapies with preclinical promise actually translate into useful therapies. They’re simply evolving with science, as they must under the “selective pressure” of advances in technology and understanding of biology.
Source